



Is My Product Regulated by the FDA or EPA?
Whether developing a product that can be used to disinfect household surfaces, a sterilizer used for medical devices, or an air purifier intended for medical purposes, it is important to first understand the regulatory framework that governs the manufacture, import, and sale of these products in the U.S.
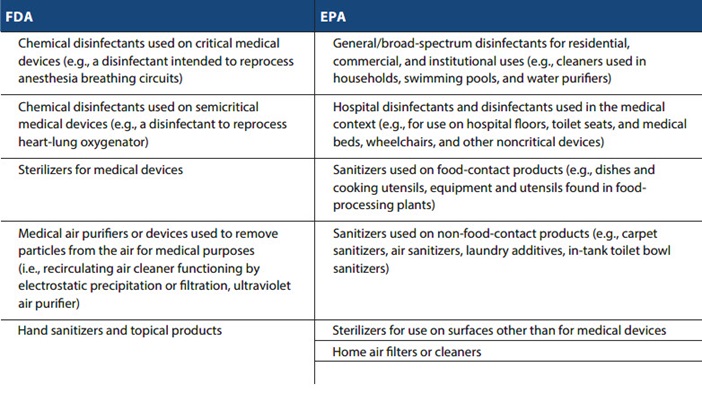
Chemical disinfectants
Chemical disinfectants are regulated by either the FDA or EPA, depending on the intended use of the disinfectant.
FDA: Under the Federal Food, Drug, and Cosmetic Act (FDCA), the FDA regulates devices that include an “instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory” that is intended to cure, mitigate, treat, or prevent disease or is intended to affect the structure of any function of the body. Under this authority, the FDA regulates chemical disinfectants used on articles that meet the definition of “device” and accessories to these devices. As reflected in the Memorandum of Understanding Between the FDA and the EPA on the Regulation of Liquid Chemical Germicides Intended for Use on Medical Devices (MOU), the FDA’s jurisdiction is limited to those sterilants/high-level disinfectants intended for use to reprocess reusable, heat-sensitive critical or semicritical medical devices. For example, heart-lung oxygenators and laparoscopes are considered “critical,” while anesthesia breathing circuits, endotracheal tubes, and cystoscopes are considered “semicritical” devices.
EPA: The EPA regulates chemical disinfectants and sanitizers that are used on inanimate objects and hard surfaces (as opposed to those used on humans or critical and semicritical medical devices) under the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA). These disinfectants and sanitizers are referred to as “antimicrobial pesticides.” The EPA’s authority over antimicrobial pesticides extends to “general use” disinfectants (e.g., for use in households, swimming pools, water purifiers) and sanitizers for both food-contact products (on sites where consumable food products are placed) and non-food-contact products (e.g., air sanitizers, carpet sanitizers, laundry additives, in-tank toilet bowl sanitizers). In the health care arena and as reflected in the MOU, the EPA’s authority over general purpose disinfectants includes disinfectants used to process “noncritical devices,” such as wheelchairs, medical beds, stands, medical lamps, and medical equipment surfaces.
Sterilizers
The FDA regulates sterilizers that are used to sterilize (or make free from viable microorganisms) medical devices. Other sterilizers outside the medical device context used to eliminate or destroy viruses are regulated by the EPA (e.g., sterilizers used on hard contact surfaces in personal care manufacturing).
Air purifiers
The FDA regulates air purifiers intended for medical purposes that are used to destroy bacteria in the air by exposure to UV radiation or remove particles from the air through filtration or electrostatic precipitation. Those same air purifiers intended for non-medical purposes, however, are regulated by the EPA. Other air cleaners or filters that kill, inactivate, entrap, or suppress the growth of viruses are regulated by the EPA. Additionally, California regulates ozone produced by electronic air cleaners for ozone production through testing and certification.
Hand sanitizers and topical products
Hand sanitizers and other topical products (e.g., hand wipes) with therapeutic purposes, such as to kill germs on the skin, are considered drugs regulated by the FDA.
The following summary chart provides examples of products regulated under either the FDA or the EPA:
How Have the FDA and EPA Traditionally Regulated These Products?
FDA-regulated products
Traditionally, manufacturers of FDA-regulated sterilizers, disinfectants, and air purifiers are required to submit a marketing application to the FDA for clearance or approval to market these products or to modify the intended use or, in certain cases, the functionality of previously cleared or approved devices. Premarket approval of a medical device requires a demonstration that the device is safe and effective when used. Within this regulatory framework, the FDA classifies medical devices based on risk and novelty of the device and its intended use. Medical devices are classified into Class I, II, and III, and regulatory clearance or approval depends on the device type in question. Most Class I devices are exempt from premarket notification under Section 510(k) of the FDCA because they are low risk. Devices that are moderate risk, i.e., because they require special controls, are classified as Class II devices and require premarket notification and clearance before market entry. To obtain clearance, the submitter of the 510(k) notification must demonstrate that the device is “substantially equivalent” to a previously cleared device. A premarket approval application (PMA) is required for devices that are high risk and for which safety and effectiveness have not been established. These devices are classified as Class III devices.
Traditionally, manufacturers of over-the-counter (OTC) hand sanitizers and other topical products may market products without FDA review or pre-approval. This does not mean that there are no requirements, however. These firms should confirm adherence to any applicable tentative final OTC monographs or deferral letters and manufacture products according to good manufacturing practices. The FDA facility registration and product listing in the FDA Drug Registration and Listing System is also required.
EPA-regulated products
The EPA regulates the distribution, sale, and use of pesticides, and defines those terms broadly. If you offer your product for sale, hold your product pending sale, ship your product, or offer to deliver your product, you have distributed and sold that product under EPA rules. It is critical to understand whether the EPA will consider your product to be a pesticide. Pesticides are substances intended to prevent, destroy, repel, or mitigate a “pest,” and a pest is defined broadly under FIFRA. It includes insects, weeds, plants, viruses, bacteria, and other microorganisms, except those living on humans or animals. The definition of pesticide turns on “intent,” and the EPA determines intent by examining claims on the label and in advertising, and the composition, mode of action, and use of the product.
If your product is a pesticide, you are prohibited from distributing or selling your product without getting an EPA registration to do so. The EPA has strict rules for what you must say about your product on its packaging and what you cannot say about it, even on your website. A pesticide must also be manufactured in an EPA-registered establishment, and the packaging must include that number on the packaging for your product. Even pesticides imported from overseas must be produced in an EPA-registered establishment, and your shipment importing the product into the U.S. will be detained and often refused if you do not have the establishment number of your packaging. The EPA and Customs and Border Protection work together to enforce these requirements for any pesticide imports. You are required to submit a notice of arrival to the EPA before your shipment arrives, and Customs will not release your shipment until the EPA gives them the green light.
Products for both FDA- and EPA-regulated uses
Products can be marketed for both FDA- and EPA-regulated uses. In this circumstance, manufacturers should consider including a clear distinction delineating both FDA-cleared or FDA-approved indications from EPA-cleared indications, or “split” their product labels to include only EPA-regulated claims for their pesticide products and should state only FDA-regulated claims for their medical devices.
What FDA Requirements Have Changed?
On March 29, 2020, the FDA published an enforcement policy, effective immediately, aimed to increase availability of FDA-regulated sterilizers, disinfectant devices, and air purifiers. This policy applies to specific products used to sterilize or disinfect medical equipment and reduce the risk of viral exposure for patients and health care providers. To aid manufacturers, the enforcement policy includes applicable product codes and classifications for each product category. Importantly, the scope of this policy does not apply to sterilizers, disinfectant devices, and air purifiers that are intended to prevent or reduce the risks of hospital-acquired infections or COVID-19.
Under the FDA’s enforcement policy, under two specific scenarios, the agency has announced it does not intend to object when the manufacturer fails to comply with the following regulatory requirements but the device nonetheless does not create an “undue risk”:
- Prior submission of a premarket notification under Section 510(k) of the FDCA and 21 CFR 807.81.
- Submission of a PMA Supplement under Section 515 of the FDCA and 21 CFR 814.39.
- Registration and Listing requirements in 21 CFR 807.
- Unique Device Identification requirements in 21 CFR 830 and 21 CFR 801.20.
The first scenario involves “limited modifications” to the indications or functionality of FDA-cleared or approved products covered by the enforcement policy “pertaining to a device’s virucidal effectiveness against SARS-CoV-2.” The FDA specifically notes that this policy would apply to a manufacturer that had previously received 510(k) clearance for a steam sterilizer intended for sterilization of medical devices when the manufacturer intends to include a statement in the labeling that the device is effective in killing SARS-CoV-2. Note that the enforcement policy does not provide any further clarity regarding what the FDA considers a “limited modification” in this scenario but addresses modifications in the following scenario.
The second scenario involves new sterilizers, disinfectants, or air purifiers that are intended to be effective at killing the SARS-CoV-2 virus and that do not have an FDA marketing authorization. As an example, this would apply to a manufacturer of a new medical air purifier that has not been approved or cleared by the FDA and that is effective in filtering out dust particles and bacteria, and the manufacturer would like to modify the filter mesh size to filter out viruses, including the SARS-CoV-2 virus. This could also apply to a manufacturer of a new sterilizer that is effective in killing the SARS-CoV-2 virus but has not otherwise been approved or cleared by the FDA.
To ensure devices do not create an “undue risk,” the FDA has outlined specific performance and labeling requirements that must be met. Importantly, manufacturers must document any changes made to the device in the manufacturer’s device master record and change control records “and make this information available to FDA, if requested, consistent with 21 CFR 820.30 and 21 CFR 820.180.” Manufacturers of all Class II, Class III, and specific Class I devices listed under 21 CFR 820.30(a)(2) must establish and maintain procedures to control the design of the device ensuring that specified design requirements are met. Under 21 CFR 820.30, manufacturers must establish and maintain a design history file containing or referencing the records that demonstrate that the design was developed in accordance with the approved design plan and the requirements under that section. Therefore, under the enforcement policy, any changes to the design or performance of a device would need to be reflected in the design history file.
Performance requirements
For sterilizers, disinfectant devices, and air purifiers under the FDA’s jurisdiction, the FDA’s enforcement policy provides recommendations for the design, evaluation, and validation of performance. The agency invites firms to discuss any alternative to the recommendations with the FDA.
- Sterilizers: Modifications, including changes to the indications or functionality, to sterilizers and their accessories should be designed, evaluated, and validated in accordance with FDA-recognized standards, which are specifically included for steam sterilizers, dry heat sterilizers, ethylene oxide sterilizers, other sterilizers, chemical indicators, sterile packaging, rigid sterilization containers, and biological indicators.
- Disinfectant devices: The FDA recommends that modifications to disinfectant devices be performed in accordance with the listed standards in the enforcement policy. When combined with typical cleaning/reprocessing practices, the FDA also recommends that manufacturers evaluate whether products meet the level of disinfection consistent with its intended use and labeling. Further, the FDA provides specific performance recommendations for low-, intermediate-, and high-level disinfection, and the agency also includes recommendations if the device includes a UV lamp or if the device generates ozone.
- Air purifiers: The FDA includes a number of performance recommendations, including, among other things, demonstration of a 4 log reduction of claimed particulates and effectiveness against a representative virus if intended for use related to SARS-CoV-2.
Labeling requirements
In addition to performance requirements, the FDA’s enforcement policy includes recommended labeling guidelines to help users better understand the device modifications. This includes the following:
- “A clear description of the available data on the device’s new indications or functions related to SARS-CoV-2 or co-existing conditions, such as: (a) device performance; and (b) potential risks (e.g., risk of UV exposure).”
- “A clear distinction delineating FDA-cleared or FDA-approved indications from those that are not FDA-cleared or FDA-approved. In addition, FDA recommends the labeling include a general statement about changes that have not been cleared by FDA.”
- “For all disinfectant devices, a clear statement of the level of disinfection.”
- Specific recommendations for UV disinfecting devices.
Compliance considerations
Ultimately, with the issuance of its enforcement policy, the FDA has effectively lowered the standard for market entry for products covered under the policy, requiring that instead of ensuring safety, the device must not create an undue risk, which is demonstrated by following the FDA’s recommendations on performance requirements and labeling practices. On labeling, manufacturers should review the FDA’s guidelines in the enforcement policy.
Typically, the FDA enforces compliance with these types of recordkeeping requirements through establishment inspections. However, without requiring that manufacturers register with the agency and submit a device list, under the enforcement policy, absent a medical device report regarding a product’s safety, the FDA may have difficulty identifying manufacturers that are introducing noncleared or unapproved devices into interstate commerce to enforce the policy’s requirement to make the device’s master record and change controls records available to the FDA, if requested. Notwithstanding this, outside the enforcement policy, the FDA lacks the authority to request records to effectuate the language included in the policy. Therefore, without the requisite enforcement authority, device companies may be able to refuse to produce such records to the agency.
For hand sanitizers, the FDA recently issued a temporary guidance for manufacturing alcohol-based sanitizers, for compounding alcohol-based hand sanitizer products, and for manufacturing alcohol to be used in hand sanitizer products. The FDA is not intending to take action against firms manufacturing alcohol-based hand sanitizers intended for use by consumers and health care professionals, so long as they: (1) are registered with the FDA; (2) list products in the FDA Drug Registration and Listing System; (3) meet specific manufacturing and labeling conditions; (4) document key steps and controls to ensure that preparation of the sanitizer under each batch matches the formula; (5) prepare the sanitizer under sanitary conditions and utilize equipment that is maintained and fit for that purpose; and (6) use the most accurate method of analysis available to confirm alcohol content.
What EPA Requirements Have Changed?
In an emergency, the EPA can authorize limited use of unregistered pesticides and pesticides registered for other uses. The EPA did just that on January 29, 2020, when it activated its Guidance to Registrants: Process for Making Claims Against Emerging Viral Pathogens Not on EPA-Registered Disinfectant Labels (EVP Guidance). The EVP Guidance provides a two-step process to allow the use of EPA-registered disinfectants against emerging viral pathogens not included on the product label. First, a registrant must submit (and can do so before an outbreak) a request to control a specific emerging viral pathogen and add a statement to the master label and terms to its registration. When approving this label amendment, the EPA specifies how the designated statement will be published and communicated. Once an outbreak occurs, registrants with approved label amendments will be allowed to use the statement in off-label communications about the disinfectant’s use against the emerging viral pathogen.
Pursuant to the EVP Guidance, the EPA is performing expedited reviews of emerging viral pathogen label claims for already registered surface disinfectants that do not require review of additional efficacy data. To request this fast-track review, a registrant needs to submit to the EPA a description of how the product meets the eligibility criteria for use against one or more categories of viral pathogens, the viruses from the product label supporting the claim, and the supporting study ID number. A few additional items are also required, as outlined on the EPA’s website, including a revised master label. Once approved, a company can make off-label emerging viral pathogen claims as specified in the EVP Guidance.
The EPA adds approved products to List N: Disinfectants for Use Against SARS-CoV-2. Not all disinfectants on this list have been specifically tested for use against SARS-CoV-2 but are deemed qualified for use. The EPA expects these listed disinfectants to be effective against SARS-CoV-2 because they have (1) demonstrated efficacy against a harder-to-kill virus; (2) qualified for the emerging viral pathogens claim; or (3) demonstrated efficacy against another human coronavirus similar to SARS-CoV-2. On April 2, 2020, the EPA revised List N to include products on its List G: Products Effective Against Norovirus and List L: Products That Meet the CDC Criteria for Use Against the Ebola Virus because the EPA has determined that these products also meet the above criteria. The EPA has also revised List N to include the specific types of surfaces products can be used on, as well as use sites.
The EPA has recently taken a number of actions relevant to manufacturers of general disinfectants for use against SARS-CoV-2. On March 26, 2020, in an effort to increase availability of disinfectant products for use against SARS-CoV-2, the EPA announced it is allowing manufacturers of disinfectants to obtain certain inert ingredients (or inactive ingredients) from different suppliers without checking with the agency for approval. The EPA’s list of commodity inert ingredients is available here. The EPA took further action to help ease the production and availability of EPA-registered disinfectants in its March 31, 2020 announcement temporarily allowing manufacturers of disinfectants on its List N to obtain certain active ingredients from any source of suppliers without checking with the agency first.
Compliance considerations
The EPA issued an enforcement memo, COVID-19 Implications for EPA’s Enforcement and Compliance Assurance Program on March 26, 2020 detailing the agency’s plan to exercise enforcement discretion for enforcement and compliance situations related to COVID-19, including noncompliance with monitoring activities and settlement and consent decree reporting obligations and milestones. This policy does not apply to pesticides/disinfectants produced in the U.S. or imported into the U.S. The EVP Guidance also does not eliminate the need for manufacturers to comply with FIFRA. As the EPA explains on its website, the EPA and its state partners are monitoring products imported to the U.S. and points of distribution within the U.S. and taking enforcement action against companies making false claims that their disinfectants work against SARS-CoV-2.
The EPA issued a press release on April 3, 2020 explaining that protecting consumers against products with fraudulent claims of effectiveness against the coronavirus is a high priority of the agency. The EPA is using tips and complaints, as well as conducting research, to identify disinfectants that have not been registered by the EPA and is focused on products advertising antiviral, antibacterial, disinfectant, sterilizing, or sanitizing properties that claim to eliminate the virus or reduce its spread. These products are illegal and cannot be imported, distributed, or sold in the U.S. The EPA typically takes enforcement action against products such as these through stop-sale orders and penalty actions. The EPA recently issued a warning to pesticide producers, manufacturers, distributors, and importers that claim their products can be used to combat the coronavirus. The EPA will be looking for products that make claims that they can be used against the SARS-CoV-2 virus, and the agency will scrutinize them to see if they:
- Are authorized to make such a claim.
- Make additional claims that are not substantiated with data.
- Were manufactured in an EPA-registered establishment and include the establishment number on the packaging.
- Include any claim or description that is false or that could mislead someone.
The EPA has already taken enforcement actions to stop companies from importing or selling products that fail to comply with all these requirements, and companies should comply with all applicable requirements for pesticide products, including prohibitions on false and misleading claims.
Conclusion
As the COVID-19 pandemic evolves, an understanding of applicable FDA and EPA regulatory requirements is a necessity for any business that is thinking about developing, manufacturing, importing, or selling a product that can be used against the SARS-CoV-2 virus. Alston & Bird’s FDA and Environment, Land Use, and Natural Resources teams have years of experience helping clients navigate the two different regulatory programs and are closely monitoring FDA and EPA actions as the agencies continue to look for ways to increase availability of these products.
Alston & Bird has formed a multidisciplinary task force to advise clients on the business and legal implications of the coronavirus (COVID-19). You can view all our work on the coronavirus across industries and subscribe to our future webinars and advisories.